
mTORC1 and epithelial development and tumorigenesis
mTOR (mammalian target of rapamycin) is a serine/threonine kinase which exists in two complexes, mTORC1 and mTORC2. Decades of studies, conducted largely in non-epithelial or cancer cell line systems, have detailed the complex molecular mechanisms by which mTORC1 integrates growth factor and nutrient inputs to regulate the intertwined processes of cell growth, proliferation and metabolism. Yet, despite a wealth of in vitro biochemical data in cell lines, the cellular effects of mTORC1 signaling on epithelial tissue development and homeostasis have only recently started to come to light. Our laboratory has been the first to elucidate the function of the interdependent PI3K and mTORC1 signaling pathways in epithelial morphogenesis. Focusing on prostatic morphogenesis, we demonstrated that basal mTORC1 signaling inhibits branching in urogenital sinus organ cultures, likely by feedback to upstream growth factor receptors(PI3K/mTOR Signaling Regulates Prostatic Branching Morphogenesis). In the murine mammary gland, constitutive mTORC1 signaling (via conditional TSC1 or PTEN deletion) is sufficient to inhibit branching in vivo and in a 3D organotypic culture system (mTOR Signaling Feedback Modulates Mammary Epithelial Differentiation and Restrains Invasion Downstream of PTEN Loss). Mechanistically, we demonstrated that mTORC1 activation restrains mammary epithelial invasion via negative feedback on EGFR/SRC signaling. Intriguingly, we also found that mTORC1 regulates the epithelial differentiation program during mammary development. These studies were the first to uncover a role for mTORC1 and mTORC1-modulated feedback in epithelial morphogenesis and differentiation. However, the relative complexity of the prostatic and mammary systems has made it difficult to tease out the detailed molecular mechanisms involved. In order to address these issues, we have exploited what is arguably the best studied mammalian system of epithelial development, the murine epidermis. Using a number of mouse models we have developed to specifically perturb mTORC1 activity during epidermal development, we have recently demonstrated that mTORC1 signaling in the epidermis is required for desmosomal cell-cell adhesion and keratinocyte biochemical differentiation, as exemplified by epidermal blistering, defective skin barrier function, and neonatal lethality of the conditional knockout mice. Mechanistically, these findings are a direct consequence of heightened cytoskeletal tension due to increased ROCK activity, resulting in impaired desmosomal gene expression and adherens junction maturation. (mTORC1 loss impairs epidermal adhesion via TGF-β/Rho kinase activation). Currently, we are addressing additional questions related to these findings, including:
What are the transcription factors mediating desmosomal gene regulation in mTORC1 loss of function keratinocytes? What are the effectors of ROCK mediating these effects?
 Rheb-cKO pups are smaller at P0, with thin, shiny skin, and fail to exclude toluidine blue dye at E18.5 consistent with an epidermal barrier defect
Rheb-cKO pups are smaller at P0, with thin, shiny skin, and fail to exclude toluidine blue dye at E18.5 consistent with an epidermal barrier defect




Another focus in the lab has been on investigating how cancer cells maintain lysosomal content in the face of persistent mTORC1 signaling. The MiT/TFE (micropthalmia-associated transcription factor) family of transcription factors consists of four conserved family members (MITF, TFE3, TFEB, TFEC) that are essential regulators of autophagy, lysosomal biogenesis, cellular energy and homeostasis and negatively regulated by mTORC1. mTORC1 is a key oncogenic signaling pathway, yet high levels of MiT/TFE activity, lysosomal biogenesis and autophagy must coexist during tumor progression to enable cells to endure nutrient-poor conditions and facilitate metastases. Strikingly, however, we have recently found that constitutive mTORC1 activation due to Tsc1 loss in murine epidermis/ keratinocytes drives the expression and nuclear localization of these TFs and increases lysosomal biogenesis. This paradoxical increase in lysosomal biogenesis by mTORC1 was mediated by feedback inhibition of AKT, and a resulting suppression of AKT-induced MiT/TFE downregulation (mTORC1 feedback to AKT modulates lysosomal biogenesis through MiT/TFE regulation). Currently, we are translating these exciting findings to understand the role of MiT/TFEs and mTORC1 function in mouse and human renal cell carcinoma due to TSC1/2 loss.
 Lysosomal genes/proteins are up-regulated with Tsc1 loss
Lysosomal genes/proteins are up-regulated with Tsc1 loss


Translational Studies in Prostate Cancer
PTEN
The translational research program in our lab has centered on the analytic, pre-analytic and clinical validation of a number of tissue-based biomarkers in prostate cancer which are useful for molecular classification of prostate tumors and which may serve as prognostic and/or predictive biomarkers.
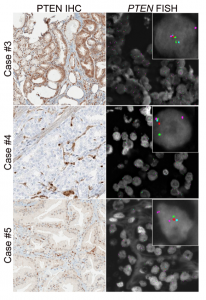
Prostate cancer cases showing variable PTEN protein expression with hemizygous PTEN gene deletion
We are best known for our work on PTEN, the most commonly lost tumor suppressor in prostate cancer and the most prevalent molecular alteration associated with patient outcomes in the disease (Ahearn et al, JNCI, 2016, Lotan et al, Mod Pathol, 2016, and Lotan et al, Oncotarget, 2017). Working collaboratively with national and international groups, we completed genetic validation of this assay in more than 5000 prostate tumors, and it is now used in CLIA-accredited laboratories and routinely in clinical surgical pathology at Johns Hopkins. We have established this assay as one of the most highly validated diagnostic and prognostic biomarkers in prostate cancer. With the recent advent of potent AKT inhibitors for the treatment of metastatic prostate cancer, we are now focused on use of the assay as a predictive biomarker in the setting of a number of completed and ongoing clinical trials. We have obtained CTEP approval and are currently testing PTEN in CHAARTED (ECOG3805) and RTOG9601, two of the largest contemporary practice-changing clinical trials in prostate cancer. We are also currently working on a gene expression signature for PTEN loss in prostate cancer.
A Prospective Investigation of PTEN Loss and ERG Expression in Lethal Prostate Cancer
Analytic Validation of a Clinical-Grade PTEN Immunohistochemistry Assay in Prostate Cancer by Comparison to PTEN FISH
TP53, RB1, AR-V7 and DNA repair proteins
In parallel, we have also worked on highly validated assays to assess additional tumors suppressors, including TP53 and RB1 mutational status in prostate cancer. We were the first group to demonstrate that these tumor suppressors are key drivers of neuroendocrine prostate cancer and can be used as predictive biomarkers in this context, since neuroendocrine tumors do not respond to androgen deprivation therapy and must be treated with chemotherapy (Tan et al, Clin Cancer Res, 2014; Tsai et al, Clin Cancer Res, 2015). Furthering this interest, we have validated additional predictive biomarkers, such as testing for mismatch repair gene defects in prostate cancer, RNA in situ hybridization for AR-V7, the androgen receptor splice variant associated with resistance to novel AR-targeted therapies in prostate cancer (Guedes et al, Clin Cancer Res, 2016—cover article) and a gene expression signature for neuroendocrine differentiation in prostate cancer (Tsai et al, BMC Cancer, 2017). Currently we are working on novel assays for constituents of the homologous DNA repair pathway which function as predictive biomarkers in prostate cancer.
Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma
Cyclin D1 Loss Distinguishes Prostatic Small Cell Carcinoma from Most Prostatic Adenocarcinomas
 Heterogeneous p53 immunostaining on standard histologic sections was common in primary tumors with discordant TP53 sequencing and p53 immunostaining results
Heterogeneous p53 immunostaining on standard histologic sections was common in primary tumors with discordant TP53 sequencing and p53 immunostaining results

African-American Prostate Cancer Biology
Another interest of our lab is the role of racial ancestry in prostate tumor molecular classification and racial disparities in prostate cancer outcomes. We were among the first to demonstrate that PTEN loss is significantly more infrequent among patients of African-American descent compared to European-Americans, suggesting that there are underlying biological differences between prostate tumors in each group (Tosoian, Eur Urol, 2017; Kaur et al, Mod Path 2018; Kaur et al, Human Path, 2019). This was particularly surprising since African-American patients with prostate cancer are 2.5 times more likely to die of the disease than their European-American counterparts. Subsequently, we led one of the largest sequencing efforts for primary prostate tumors in African-American patients (Faisal, Clin Can Res, 2020, in press). Our lab is a key member of the recently NCI-funded RESPOND study which will collect and molecularly characterize the largest prospective cohort of African-American prostate cancer cases (http://respondstudy.org/).
 Immunostaining for CD3, CD8, and FOXP3 identifies respective specific subsets of tumor-infiltrating lymphocytes in prostate tumor
Immunostaining for CD3, CD8, and FOXP3 identifies respective specific subsets of tumor-infiltrating lymphocytes in prostate tumor
